stock here: its always good to look back at research done “before everyone had a horse in the race”. I think I found one of the smoking guns to support Robert Kennedy’s Assertion greater deleterious effects on Whites and Blacks, and less effects on Jews and Finns.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7360473/

So now Bobby Kennedy is saying these cleavage sites works different on different races.
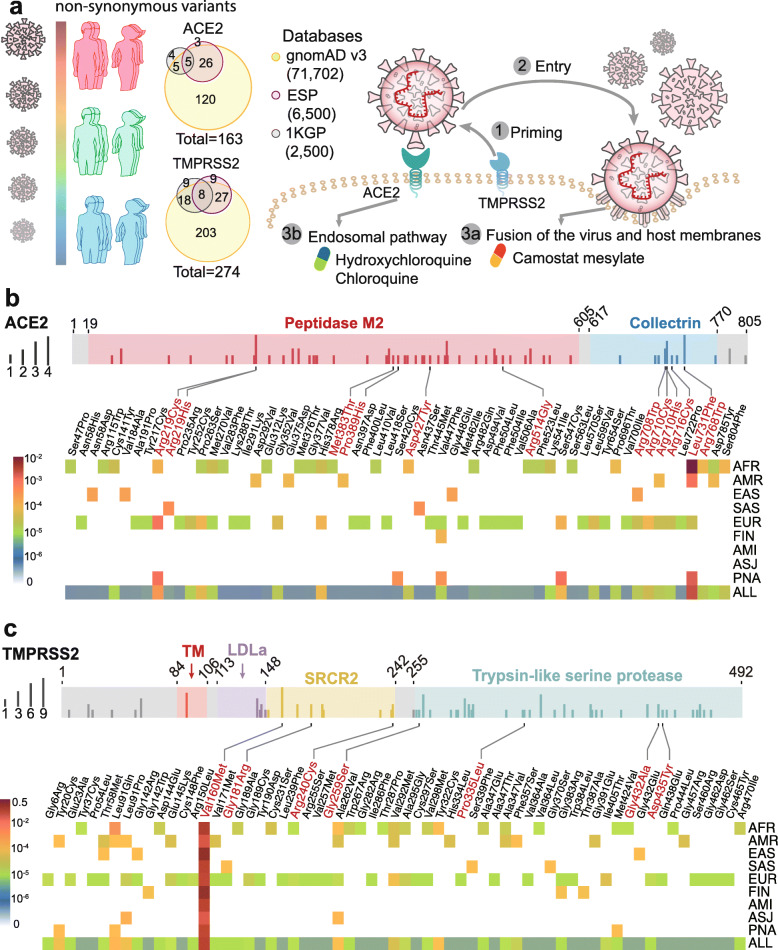
The coding-region variants in ACE2 and TMPRSS2 from ~ 81,000 human genomes across 8 populations. a Coding-region variants in the genes encoding angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) across three human genome databases: (i) Genome Aggregation Database (gnomAD v3), (ii) Exome Sequencing Project (ESP), and (iii) 1000 Genomes Project (1KGP). SARS-CoV-2 utilizes the host cell factors angiotensin-converting enzyme 2 (ACE2) for entry into cells and the host transmembrane serine protease TMPRSS2 for SARS-CoV-2 spike (S) protein priming, offering potential pathway for therapeutic development in treatment of COVID-19. b Distribution of 61 deleterious variants in the ACE2 coding region identified in gnomAD (v3). Polyphen2 > 0.96 and CADD scores > 20 as cutoff identify putative deleterious variants. The upper panel using 3 colors shows the functional domains of ACE2, and the height of the vertical line represents the number of populations that carry this variant. The lower heatmap shows the allele frequencies (color key) of a variant across different populations. c Distributions of 63 putative deleterious variants in the TMPRSS2 coding region using the same approach of b. AFR, African/African-American; AMI, Amish; AMR, Latino/Admixed American; ASJ, Ashkenazi Jewish; EAS, East Asian; FIN, Finnish; EUR, Non-Finnish European; SAS, South Asian; PNA, population not assigned
ACE2 polymorphism analysis across different populations
Here, we investigated genetic susceptibility to COVID-19 by examining DNA polymorphisms in ACE2 (OMIM 300335) and TMPRSS2 (OMIM 602060) genes. We assembled a total of 437 non-synonymous single-nucleotide variants (SNVs) in the protein-coding regions of ACE2 and TMPRSS2 (Fig. 1a) from three databases: (i) Genome Aggregation Database (gnomAD v3: gnomad.broadinstitute.org, covering 9 geographical areas), (ii) Exome Sequencing Project (ESP: evs.gs.washington.edu/EVS/), and (iii) 1000 Genomes Project (1KGP, www.internationalgenome.org). We used ANNOVAR [9] to annotate all non-synonymous variants. By applying Polyphen2 and CADD (Combined Annotation Dependent Depletion) scores, we identified 63 potentially deleterious variants in ACE2 (61 in gnomAD) and 68 deleterious variants in TMPRSS2 (63 in gnomAD).
We found that the distribution of deleterious variants in ACE2 differs among 9 populations in gnomAD (v3). Specifically, 39% (24/61) and 54% (33/61) of deleterious variants in ACE2 occur in African/African-American (AFR) and Non-Finnish European (EUR) populations, respectively (Fig. 1b). Prevalence of deleterious variants among Latino/Admixed American (AMR), East Asian (EAS), Finnish (FIN), and South Asian (SAS) populations is 2–10%, while Amish (AMI) and Ashkenazi Jewish (ASJ) populations do not appear to carry such variants in ACE2 coding regions (Fig. 1b). Specifically, several variants, including p.Met383Thr, p.Pro389His, and p.Asp427Tyr, have been reported to slightly inhibit the interaction between ACE2 and the spike protein of SARS-CoV-1 [10], which caused the first global SARS-CoV-1 outbreak. Only AFR populations carry p.Met383Thr and p.Asp427Tyr variants, with allele frequencies of 0.003% and 0.01%, respectively (Fig. 1b). The p.Pro389His only occurs in the AMR populations, with an allele frequency of 0.015%. The p.Arg514Gly is a low allele frequency (0.003%) variant in AFR populations and is also somatically mutated in colon cancers and melanomas from The Cancer Genome Atlas (TCGA: https://portal.gdc.cancer.gov). This ACE2 variant is located in the angiotensinogen (AGT)-ACE2 interaction surface, which is anticipated to influence the renin-angiotensin system (RAS) function. The RAS is critical for regulation of blood pressure, sodium, and fluid balance, and its dysfunction is associated with cardiovascular and kidney disorders [11]. Residues Arg708/710/716 are located in the dimeric interface of ACE2 (Fig. 2a), and they are essential for its cleavage by TMPRSS2; this processing is required for augmentation of SARS-S-driven entry into host cells [12]. The EUR population carries the p.Arg708Trp, p.Arg710Cys, p. Arg710His, and p.Arg716Cys variants with allele frequency of 0.01~0.006% (Fig. 1a), while the EAS and the AMR populations only carry p.Arg708Trp and p.Arg710His with allele frequency of 0.04% and 0.01% respectively. In addition to these four variants, p.Leu731Phe has the highest allele frequency in the AFR and EUR populations. We further inspected the expression quantitative trait loci (eQTL) for ACE2 using the GTEx [13] and QTLbase [14] databases. We did not find any eQTLs for ACE2 from the GTEx, while we found one weak eQTL associated with ACE2 non-synonymous SNP (rs41303171) in the kidney from the QTLbase [14].
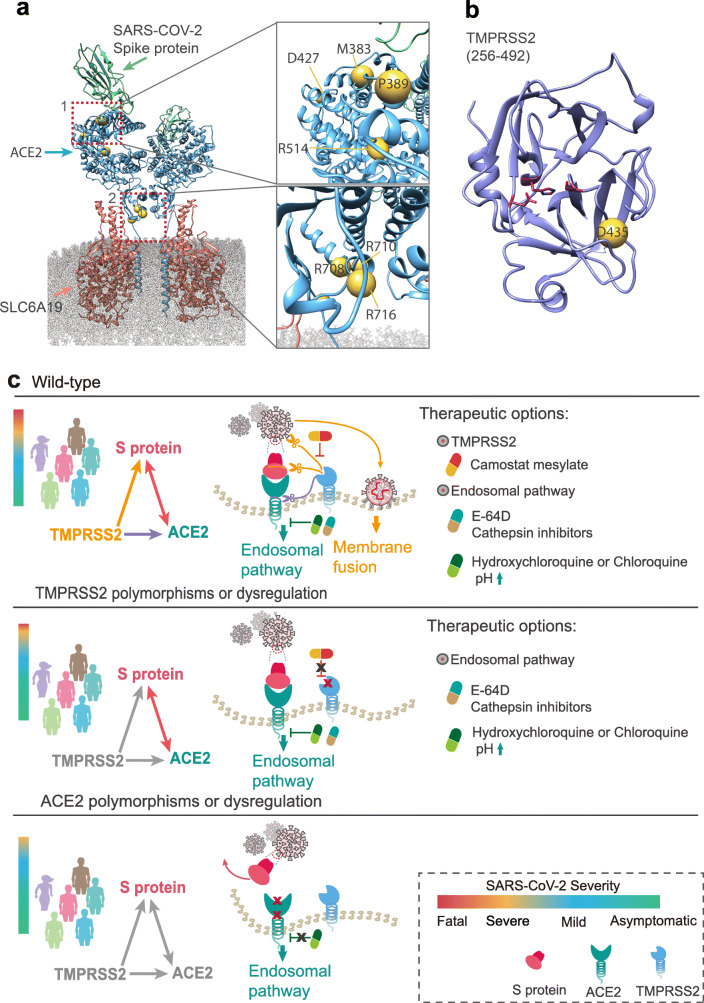
Structural view of the coding-region variants in ACE2 and TMPRSS2 and a proposed pharmacogenomics model of effective combination therapies for COVID-19. a Full-length structures of the sodium-dependent neutral amino acid transporter B(0)AT1 (SLC6A19, red)–ACE2 (blue) heterodimer in its homodimeric form complexed with the receptor binding domain (RBD, mint) of SARS-CoV-2 (PDB ID: 6M17). Highly deleterious variants are labeled as yellow spheres on ACE2. Insets depict mutations in residues 383 through 427 (top) and residues 708 through 731 (bottom). b Homology model of the catalytic chain (256–492) of TMPRSS2. Highly deleterious mutations are labeled as yellow spheres. c A proposed model of effective combination therapies (i.e., hydroxychloroquine, E-64D (a protease inhibitor), and camostat mesylate (an approved TMPRSS2 for treatment of chronic pancreatitis in Japan)) for COVID-19 by blocking ACE2 and TMPRSS2 across different populations with three genotypes. Relationship among spike (S) protein of SARS-CoV-2, ACE2, and TMPRSS2 were shown as a triangle, with each pair connecting by physical binding (double-headed arrow) or cleavage (single-headed arrow). We propose three hypotheses for COVID-19 therapeutic options: (i) for patients with wild-type or naïve expression of ACE2 and TMPRSS2, hydroxychloroquine (or chloroquine, or E-64D) combined with camostat may offer more clinical benefit; (ii) for patients with polymorphisms or dysregulation on TMPRSS2, hydroxychloroquine or chloroquine monotherapy may offer more clinical benefit; and (iii) for patients with polymorphisms or dysregulation on ACE2, the patients who might have mild symptoms can recover in a short period. All three pharmacogenomics models for COVID-19 must be validated both experimentally and clinically before being used in patients
Altogether, these comparative genetic analyses suggest that ACE2 genomic variants may play important roles in susceptibilities to COVID-19 and its associated cardiovascular conditions by altering AGT-ACE2 pathway (i.e., p.Arg514Gly). In addition to differential polymorphisms which may explain susceptibility and even outcome in different ethnic populations, the fact that ACE2 is localized to Xp22.2 may help explain the observed male-associated risk. As such, even in the absence of variation in this gene, the monoallelic versus biallelic presence of this gene may impact the natural history and prognosis of COVID-19 in males.
TMPRSS2 polymorphism analysis across different populations
TMPRSS2 enzyme activity is important for coronavirus spread and pathogenesis in the infected host [15]. Our analysis indicates 4% (11/274) of non-synonymous variants of TMPRSS2 are stop-gained mutations and carried by AFR and EUR with low allele frequency (7.0 × 10−6~1.4 × 10−5). Meanwhile, 35% (22/63) and 59% (37/63) of deleterious variants in TMPRSS2 coding regions are carried by the AFR and EUR populations from gnomAD (v3), respectively (Fig. 1c). Each of the EAS, SAS, and FIN populations only carries 4 deleterious variants. We found 6 germline deleterious variants (p.Val160Met, p.Gly181Arg, p.Arg240Cys, p.Gly259Ser, p.Pro335Leu, and p.Gly432Ala) in the TMPRSS2 coding region, which are also identified as somatic mutations occurring in different cancer types from TCGA and COSMIC databases (https://cancer.sanger.ac.uk/cosmic).
We further evaluated the eQTL profile of TMPRSS2 using the GTEx [13] and QTLbase databases [14] as well. We found two eQTLs associated with TMPRSS2 non-synonymous SNPs (rs12329760 (encoding p.Val160Met), p = 4.54 × 10−5; rs75603675, p = 0.009) in the kidney and bone, respectively, using the QTLbase database [14], while there are no known eQTLs associated with TMPRSS2 non-synonymous SNPs from GTEx [13]. Notably, all populations carry p.Val160Met variants with the highest allele frequency (~ 25%), especially for the EAS population at a 40% allele frequency. Asp435 is a key residue for catalytic substrate binding of TMPRSS2 (Fig. 2b). We found that the p.Asp435Tyr, which has low allele frequency, is carried by the EUR population only (Fig. 1c). These unique but prevalent polymorphisms in TMPRSS2 offer potential explanations for differential genetic susceptibility to COVID-19 as well as for risk factors, including those with cancer and the high-risk group of male patients. Because TMPRSS2 is located on 21q22.3, we could speculate that individuals with Down syndrome would be at high risk for COVID-19 infection. In addition, oncogenic roles of TMPRSS2 may be linked to poor outcomes with COVID-19 as well [16], which should be studied in the future. Using single-cell RNA-sequencing analysis, Schuler et al. showed that TMPRSS2 expression was highest in ciliated cells and type I alveolar epithelial cells (AT1) and increased with aging in humans and mice [17]. This observation suggests that developmental regulation of TMPRSS2 may link the relative protection of infants and children from COVID-19. Thus, it should be of great interest to investigate the age-related polymorphisms for TMPRSS2, such as using the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort [18], in the future.
Host genetic factors guide personalized treatment of COVID-19
There are currently no approved effective medications against COVID-19. Several national and international research groups are working on the development of vaccines to prevent COVID-19, but effective vaccines not likely to be available for many months. Several potentially repurposable drugs (Fig. 2c), including melatonin [19], hydroxychloroquine, and chloroquine, are under investigation for treatment of COVID-19 [20]. A primary mechanism-of-action of hydroxychloroquine and chloroquine is to inhibit virus entry by targeting the endosomal pathway [20]. Hydroxychloroquine and chloroquine is known to increase the pH of endosomes, which inhibits membrane fusion, a required mechanism for viral entry into the cell [21]. Additionally, inhibition of SARS-CoV-2 could be due to differential glycosylation of both ACE2 and the spike protein [21]. As shown in Fig. 1b, several variants identified in the AFR and AMR populations, including p.Met383Thr, p.Pro389His, and p.Asp427Tyr (the pathogenic variants in ACE2 slightly inhibit interaction with the S protein), may influence the clinical efficacy of hydroxychloroquine or chloroquine. This may help explain why treatment of hydroxychloroquine was not significantly associated with difference in in-hospital mortality [22]. However, further pharmacogenomic studies that integrate drug response and genetic data from patients with COVID-19 are urgently needed.
In addition to the endosomal pathway, fusion of viral and host cellular membranes through S protein conformational changes is another way for coronavirus entry into the host cell [23]. This process can be blocked by a TMPRSS2 inhibitor (camostat mesylate, a drug approved in Japan) [5]. The mechanisms whereby TMPRSS2 promotes cellular entry of SARS-CoV-2 can be summarized by two aspects based on its proteolytic function (Fig. 2). The first is S protein cleavage at S1/S2 and S2’ sites, which might be the reason why SARS-CoV-2 entry into cells depends on TMPRSS2. The infection and pathogenesis of SARS-CoV-2 depends on the presence of TMPRSS2, in the face of the cellular elevated pH environment [5, 24, 25]. The inhibitors of endosomal acidification such as CatB/L inhibitor E-64D and hydroxychloroquine/chloroquine may only work for TMPRSS2-absence patients who are infected by SARS-CoV-2, and may have less effect or no effect for the patients with wild-type of TMPRSS2 [5, 24]. Therefore, the EUR and AFR populations might be more sensitive to hydroxychloroquine or chloroquine by carrying missense variants and stop-gained variants on TMPRSS2 (Figs. 1c and and2c).2c). Yet, for patients who have wild-type of ACE2 and TMPRSS2, a combination of camostat with hydroxychloroquine or chloroquine may have better clinical benefit. However, all discussed treatment strategies must be validated by randomized controlled trials before clinical use. The second mechanism is cleavage of ACE2 by TMPRSS2 at Arginine 697 to 716 [12], which enhances viral uptake. Thus, the EUR population with p.Arg708Trp, p.Arg710Cys, p.Arg710His, and p.Arg716Cys variants in ACE2 may have mild symptoms after SARS-CoV-2 infection as ACE2 loses the cleavage site by TMPRSS2 and changes the ACE2 dimer formation [26] (Fig. 2c).
Discussion and future directions: call for host genetics initiative for COVID-19
A few limitations merit consideration. Current analysis examined massive genomic data from general population, not COVID-19 patient-specific populations. All genetic associations identified in current study are urgently needed to be tested in COVID-19 patients in the near future. As the high-resolution protein structure of TMPRSS2 is not yet available, further functional observations and clinical validation are warranted for all abovementioned genetic and pharmacogenomics findings. We anticipate that large-scale genome-wide association studies (GWAS) are urgently needed to identify likely causal host genetic risk factors for severe COVID-19 outcomes using genetic data from patients with COVID-19; such knowledge will improve risk stratification of individuals exposed to or testing positive for SARS-CoV-2 and allow for precision medicine interventions for COVID-19. A COVID-19 host genetics initiative is already underway to bring together the human genetics research community to generate, share, and analyze data in a search for the genetic determinants of COVID-19 susceptibility, severity, and outcomes [27]. The first COVID-19 GWAS identified the 3p21.31 gene cluster (including SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, and XCR1) as a genetic susceptibility locus in severe patients with COVID-19 and respiratory failure [28]. Yet, our study aims to look for SNPs associated with disease severity of COVID-19, but not disease susceptibility. In summary, systematic identification of the genetic determinants of COVID-19 susceptibility, severity, and clinical outcome, including both virus and host factors (e.g., ACE2 and TMPRSS2 polymorphisms), could guide personalized treatment in the emerging COVID-19 pandemic and even explain current epidemiologic observations (i.e., males, elderly at high risk, and clinical comorbidities) and natural history.
Conclusions
This comprehensive comparative genetic analysis of approximately 81,000 human genomes suggested possible associations of ACE2 and TMPRSS2 DNA polymorphisms with COVID-19 susceptibility, severity, and clinical outcomes. We found that ACE2 polymorphisms were more likely to be associated with cardiovascular and pulmonary conditions by altering the angiotensinogen-ACE2 interactions, such as p.Arg514Gly in the African/African-American population. Unique but prevalent polymorphisms in TMPRSS2, including p.Val160Met (rs12329760), may provide potential explanations for differential genetic susceptibility to COVID-19 as well as for risk factors, including cancer and the high-risk group of male patients. We highlighted that polymorphisms in ACE2 or TMPRSS2 could guide personalized treatments (i.e., hydroxychloroquine and camostat) for COVID-19. In summary, this study suggested that ACE2 or TMPRSS2 DNA polymorphisms were likely associated with genetic susceptibility to COVID-19, which calls for a human genetics initiative for fighting the COVID-19 pandemic.
Acknowledgements
We thank all helpful discussions and critical comments regarding this manuscript from the COVID-19 Research Intervention Advisory Committee members at the Cleveland Clinic. SCE is the Alfred Lerner Memorial Chair of Innovative Research and CE is the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic.
Abbreviations
| 1KGP | 1000 Genomes Project |
| ACE2 | Angiotensin-converting enzyme 2 |
| CoV | Coronavirus |
| COVID-19 | Coronavirus Disease 2019 |
| eQTL | Expression quantitative trait loci |
| gnomAD | Genome Aggregation Database |
| MERS | Middle East respiratory syndrome |
| SARS | Severe acute respiratory syndrome |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| ESP | Exome Sequencing Project |
| S | Spike |
| TMPRSS2 | Transmembrane serine protease 2 |
Authors’ contributions
F.C. conceived the study. Y.H., J.Z., and W.M. performed all experiments and data analysis. A.K., M.K.C, N.S., L.J., C.E., and S.E. discussed and interpreted all results. F.C., Y.H., C.E., and S.E. wrote and critically revised the manuscript with contributions from other co-authors. All authors read and approved the final manuscript.
Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH) under Award Number R00HL138272 and the National Institute of Aging under Award Number R01AG066707 to F.C. This work was supported, in part, by the VeloSano Pilot Program (Cleveland Clinic Taussig Cancer Institute).
Availability of data and materials
All population genetic data used in this study are free and available at three databases: (i) Genome Aggregation Database (gnomAD v3: gnomad.broadinstitute.org, covering 9 geographical areas), (ii) Exome Sequencing Project (ESP: evs.gs.washington.edu/EVS/), and (iii) 1000 Genomes Project (1KGP, www.internationalgenome.org).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The content of this publication does not necessarily reflect the views of the Cleveland Clinic. The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
1. Ashour HM, Elkhatib WF, Rahman MM, Elshabrawy HA. Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens. 2020;9(3):186. doi: 10.3390/pathogens9030186. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
2. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(5):533–534. doi: 10.1016/S1473-3099(20)30120-1. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
3. Dong Y, Mo X, Hu Y, Qi X, Jiang F, Jiang Z, Tong S. Epidemiology of COVID-19 among children in China. Pediatrics. 2020;8(6):2118–2120. [Google Scholar]
4. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
5. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. doi: 10.1016/j.cell.2020.02.052. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
6. Stopsack KH, Mucci LA, Antonarakis ES, Nelson PS, Kantoff PW. TMPRSS2 and COVID-19: serendipity or opportunity for intervention? Cancer Discov. 2020;10(6):779–782. doi: 10.1158/2159-8290.CD-20-0451. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
7. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020. 10.1007/s11684-020-0754-0. [PMC free article] [PubMed]
8. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, Wang H, Wan J, Wang X, Lu Z. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. 10.1001/jamacardio.2020.1017. [PMC free article] [PubMed]
9. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
10. Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, Wong SK, Huang IC, Xu K, Vasilieva N, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
11. Kuster GM, Pfister O, Burkard T, Zhou Q, Twerenbold R, Haaf P, Widmer AF, Osswald S. SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19? Eur Heart J. 2020;41(19):1801–1803. doi: 10.1093/eurheartj/ehaa235. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
12. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–1307. doi: 10.1128/JVI.02202-13. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
13. Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. doi: 10.1126/science.1262110. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
14. Zheng Z, Huang D, Wang J, Zhao K, Zhou Y, Guo Z, Zhai S, Xu H, Cui H, Yao H, et al. QTLbase: an integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res. 2020;48(D1):D983–D991. doi: 10.1093/nar/gkz888. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
15. Shirato K, Kawase M, Matsuyama S. Wild-type human coronaviruses prefer cell-surface TMPRSS2 to endosomal cathepsins for cell entry. Virology. 2018;517:9–15. doi: 10.1016/j.virol.2017.11.012. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
16. Yu J, Ouyang W, Chua MLK, Xie C. SARS-CoV-2 transmission in patients with cancer at a tertiary care hospital in Wuhan. China JAMA Oncol. 2020. 10.1001/jamaoncol.2020.0980. [PMC free article] [PubMed]
17. Schuler A, Habermann C, Plosa J, et al. Age-related expression of SARS-CoV-2 primining protease TMPRSS2 in the developing lung. 2020. 10.1101/2020.05.22.111187 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.111187.
18. Mostafavi H, Berisa T, Day FR, Perry JRB, Przeworski M, Pickrell JK. Identifying genetic variants that affect viability in large cohorts. PLoS Biol. 2017;15(9):e2002458. doi: 10.1371/journal.pbio.2002458. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
19. Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA. 2020. 10.1001/jama.2020.6019. [PubMed]
21. Savarino A, Di Trani L, Donatelli I, Cauda R, Cassone A. New insights into the antiviral effects of chloroquine. Lancet Infect Dis. 2006;6(2):67–69. doi: 10.1016/S1473-3099(06)70361-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
22. Rosenberg ES, Dufort EM, Udo T, Wilberschied LA, Kumar J, Tesoriero J, Weinberg P, Kirkwood J, Muse A, DeHovitz J, et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York state. JAMA. 2020. 10.1001/jama.2020.8630. [PMC free article] [PubMed]
23. Walls AC, Tortorici MA, Snijder J, Xiong X, Bosch BJ, Rey FA, Veesler D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017;114(42):11157–11162. doi: 10.1073/pnas.1708727114. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
24. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85(2):873–882. doi: 10.1128/JVI.02062-10. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
25. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
26. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
27. Initiative C-HG. The COVID-19 host genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet. 2020;28(6):715–718. doi: 10.1038/s41431-020-0636-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
28. Group TSC-G. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med. 2020. 10.1056/NEJMoa2020283. [PMC free article] [PubMed]
Articles from BMC Medicine are provided here courtesy of BioMed Central
———————————————————————————
So now Bobby Kennedy is saying these cleavage sites works different on different races.
In mid 2021, as I discovered the horrific experiment that was going on….they took the logistically insane method of sending their bad batches to very spread out states. The theory was that they were collecting data on how the specific batches affected people in different demographics, races, age, and even temperature. Shortly after publishing the undeniable data….Google banned my website on blogger, and then started erasing my history for 10 years. over the target! I had made a full backup a week or two back, I knew I was getting close to the real story and was therefore at risk.
I don’t have a firm opinion on whether “the furin cleavage site” makes effects of different races from COVID and also the Spike S protein of the Injections, in all their formulations.
I’ll add more when I get more. Research has been tough. But maybe this blood type meme will play out well.
According to the American Red Cross, the following statistics show the most common blood types in the U.S. based on the donor population:
- African American: 47% O-positive, 24% A-positive, and 18% B-positive
- Latin American: 53% O-positive, 29% A-positive, and 9% B-positive
- Asian: 39% O-positive, 27% A-positive, and 25% B-positive
- Caucasian: 37% O-positive, 33% A-positive, and 9% B-positive
Most rare blood types by ethnicity
The least common blood type in the U.S. is AB-negative, with less than 1% of the population having this type.
Statistics from the American Red Cross show that the following are the most rare forms of the major eight blood types in the U.S. based on the donor population:
- African American: 0.3% AB-negative, 1% B-negative, and 2% A-negative
- Latin American: 0.2% AB-negative, 1% B-negative, and 2% both A-negative and AB-positive
- Asian: 0.1% AB-negative, 0.4% B-negative, and 0.5% A-negative
- Caucasian: 1% AB-negative, 2% B-negative, and 3% AB-positive
—————————————————— I asked AI to help, and it appears that AI is becoming more woke….”The concept of race is a social construct” hmmmm
here it is
As of my last update in September 2021, there is no comprehensive chart that provides percentage breakdowns of blood types for different races, including the Ashkenazi Jewish population. Blood type distribution can vary widely based on geographical location, cultural background, and historical migrations. Additionally, the concept of “race” is complex and not based solely on genetic factors, so trying to associate specific blood type distributions with races can be misleading.
However, I can provide you with some general information about blood type distributions in different populations. Keep in mind that these numbers are approximate and may not be fully up-to-date:
- ABO Blood Group System:
- Blood Type O: This is usually the most common blood type worldwide.
- Blood Type A: Common in populations of European, African, and Asian descent.
- Blood Type B: Common in populations of Asian and African descent.
- Blood Type AB: Relatively rare, and often more prevalent in mixed populations.
- Rh Factor:
- Rh-positive: This is the most common Rh factor in most populations.
- Rh-negative: Relatively less common compared to Rh-positive, and its distribution can vary across different ethnic groups.
As for the Ashkenazi Jewish population, they are an ethnoreligious group with Ashkenazi Jews primarily having European ancestry. It’s important to note that their blood type distribution would likely resemble the blood type distribution of European populations.
For any specific and up-to-date data on blood type distributions in different populations, it’s best to refer to recent studies and medical databases. The distribution of blood types can also change over time due to various factors such as migration, intermixing of populations, and medical advances.
Remember, blood type information is essential for medical purposes, such as blood transfusions, but it should not be used to draw conclusions about a person’s race or ethnicity. The concept of race is a social construct and not a valid biological categorization. Genetic variation within and between populations is much more complex and nuanced than can be captured by broad racial categories.
2 replies on “Kennedy — Greater Effect on Whites and Black, Almost No Effect on Jews and Finns”
The best I can come up with is Ashkenazi Jews having about 40% type A blood, Sephardic with about 45% type “A”.
https://www.deepdyve.com/lp/wiley/blood-groups-in-ashkenazi-jews-XT40A2CJrm
I am not sure if RFK Jr. was 100% sure of it either, but in private conversation made mention of potential Gene Specific Bio-Weapons, well Governments have been working on that for a long time…
Just look at this non-comprehensive list of Jews who died by Covid:
https://www.chabad.org/generic_cdo/aid/4716515/jewish/Each-Person-a-World.htm
Even more interesting is people with type “A” blood are MORE susceptible to Covid, that would include a lot of Ashkenazi Jews:
“The A (Rh +ve) blood group seems most vulnerable, while O (Rh +ve) is least subject to the virus infection.”
https://journals.sagepub.com/doi/full/10.1177/20503121231187736
and
https://b-s-h.org.uk/about-us/news/raised-covid-risk-for-blood-type-a-explained
https://connect.mayoclinic.org/discussion/lt-covid-and-blood-type-connection/
Ya, my accountants top prinicipal was a Jew and got injected and went into the hospital later that night with a 104.5 fever. Genetic weapons are not going to be perfect.